做过分子实验的人都知道,要想做的有效率有质量是很苦逼的,1个月做100个克隆那都是小case,没点看家的本事怎么行。如果再遇到比较极品的稀有基因,周旋个把月,那也是家常便饭。下面就和大家分享一些载体构建的经验,从此迈向科研达人:
1. 准备工作
俗话说:用欲善其事,必先利其器。建议大家在做构建之前先找好工具,效果事半功倍哦。推荐两个工具,一个是oligo软件,常用于引物设计和酶切位点分析;另一个是DNAstar软件,工具极强大,一款全面的生物医学软件,用作DNA和蛋白质序列分析、重叠群拼接和基因工程管理。
插播个笑话:听说隔壁实验室里有个学医的学生来做课题,很聪明很刻苦,但除了他以外,包括老板在内都是生物物理背景,整个实验室对分子生物就只有一个粗浅的概念,这个学生就想把一个质粒上的基因插到另一个质粒上去,要是我就先查查有没有合适的酶切位点,要没有就改造一下质粒一切搞定,这学生他不懂啊(要命的是她老板固然牛,对这方面也不懂),自己辛辛苦苦设计了PCR引物去做PCR,P了将近5K的产物去测序,结果测的结果中间有个突变,要懂的就找找酶切位点,从原来的替换上去,然后测这下这段就行。他呢,又送去了若干了质粒一个接一个的测,他光测序就要花好几千(你得佩服她老板真有钱呀)。这件事教育我们准备工作是多么重要。
2. 设计引物
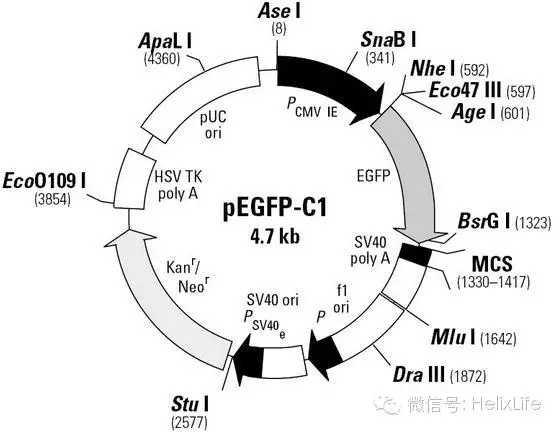
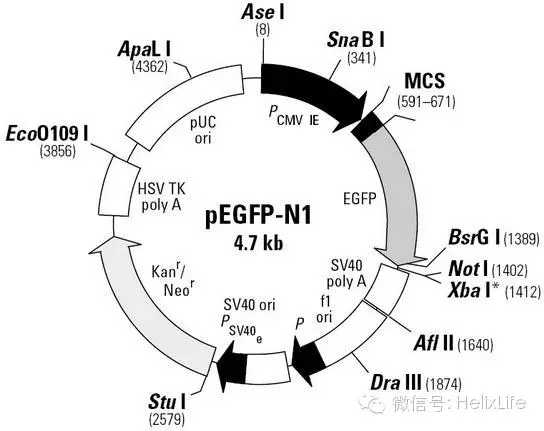
1) 注重要:看懂质粒图谱!拿大家比较熟悉的PEGFP-C1和PEGFP-N1做例子。
想用N1质粒,设计引物就得把下游引物上的中止密码子去掉,不要辛辛苦苦的一路做下来结果根本不表达融合蛋白,你就死了;C1质粒,注意阅读框,要是移码了,你也死了。而且PEGFP系列有1.2.3,要弄清楚别弄窜了。一句话,要看懂你的图谱!
其他需要注意:
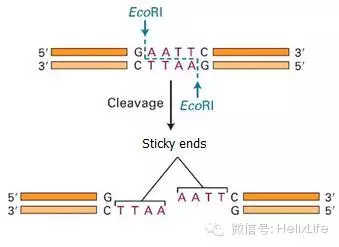
- 设计酶切位点时要加保护碱基(大家要用T载体就当我没说);
- 酶切位点设计原则:尽量用粘性末端,实在不行就用一些常用平端酶,如EcoRV和SnaB1等,要是以后还有别的用处就多加点酶切位点,曾一口气在引物上加了五个酶切位点以防以后要用到,注意计算TM值的时候要减去这些不匹配的序列;
- 注意KOZAK序列的问题,很多质粒没有提供KOZAK序列,这要在设计的时候直接在引物上加好。
- 擅用Gene Overlap,比方说加个flag标签,his标签,my标签之类的,直接设计三引物overlap一下就可以,省得还要再多构建一步,这些都是设计引物时候就要考虑好的。引物长一点不要紧(我最喜欢两步法了),尤其是对GC比高的序列,有时候引物不长PCR根本不出结果,注意如果GC比较高,这个时候Gene Overlap就不要做了,很麻烦,克隆很难挑。
- 没有合适的酶切位点?很简单,用同尾酶策略,比方说Bgl2和BamH1,Nhe1和spe1,Xho1和Sal1(注意连上了切不下来),实在不行就平端吧。
3. PCR扩增
如果没有现成的质粒可供酶切,PCR是最理想也是最方便的策略。目前市面上可选择的PCR酶实在太多,每隔一段时间还会升级,正常情况下,高保真的taq酶都能满足需求。但如果遇到难PCR的高GC基因,可以换不同PCR酶,添加一些 DMSO,甘油等,实在不行可以考虑Clontech的2 GC rich kit,价格和实力都大牛。另外,建议电泳切胶回收纯化PCR产物,去除一些非特异性条带。


4. 酶切
强烈推荐NEB的内切酶,记得有一次用别家的酶切过夜,结果质粒都被切碎了(当然当时质粒浓度也不高),而用NEB的酶,切个七十二小时仍旧一切OK,尤其现在NEB还推出了HF的内切酶,没有星活性而且统一都用Buffer4。另外TAKARA的酶也还可以。
5. 连接
最常用的是NEB的T4连接酶,很好用,但是注意Buffer里有ATP,反复冻融ATP失活很快,拿到buffer后就分装,10uL/管,一次性使用。
载体总量:一般来说,载体浓度在20-100ng/uL较好,太低的话碰撞几率低,太高的话又会产生很多非目的克隆,总量从50-100ng就可以,太低失败几率很高,太高克隆太多,挑克隆会很麻烦,反应总体积也有讲究,体积太大载体和目的片段碰撞几率太低,而且对感受态细胞也是个挑战,通用10-15uL。
载体和插入片段比例:载体和插入片段比例一般是1:7,如果很难连接,比如说平端连接,要适当提高片段浓度,同时加大比例1:10。
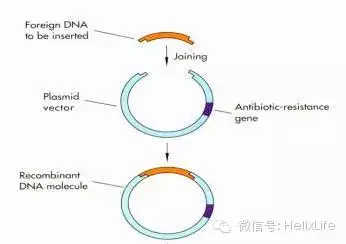
6. 转化
按照SOP做就行,话说实验室原来从takara买感受态(competent cell),后来发现还是自己做的效率高(protocol免费索取)。要注意的是LB必须无菌而且没有抗生素,实验室原来有个技术员,做一次失败一次,我就奇怪了,后来才发现他用的居然是加了kana的LB,直接晕过去了。
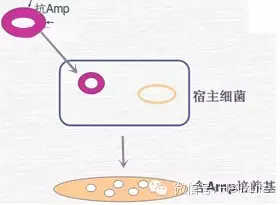
7. 鉴定
想要快速鉴定,建议用菌液PCR,菌液PCR是有一定讲究的,很多人做菌液PCR鉴定假阳性特多,为什么?因为你PCR引物选的都在载体上,注意引物必须分别在载体和插入片段上才准,PCR方法鉴定是极其准确的,数百次菌液PCR经验。PCR鉴定做起来也很简单,拿个2mL tube,装0.5mL 加入相应抗生素的LB,摇个三小时左后取1uL做模板就行了。如果想更快,直接菌落PCR也不错,效果一样。
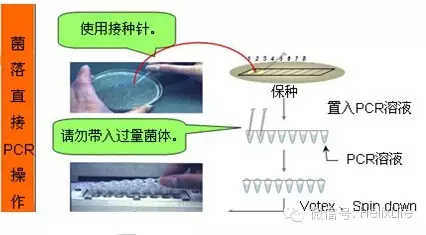
8. 测序
拿到测序结果时,不仅要核对序列,还要看测序峰图,这很重要。因为有时候光看序列对,实际上图显示的不对,或者虽然序列结果不对,但是图上出现的峰值本身就很怪异不可信,出现突变也不怕,有很大可能性是简并密码子;
还要重点检查的地方就是“接头”的地方,因为偶尔引物会出错,比方说少个碱基什么的,还有时候酶切之后连接也会丢一两个碱基什么的,如果不仔细检查到了后期悔之无及。